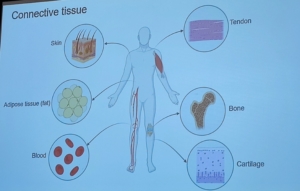
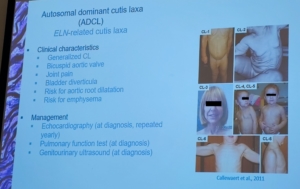
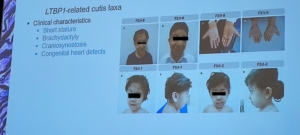
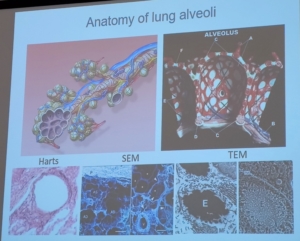
Recientemente se han descubierto 4 mutaciones genéticas nuevas:
LOX: Este gen de la familia de la 5-lisil oxidasa está implicado en el inicio de la reticulación de la elastina y el colágeno. La mutación induce síntomas cardiovasculares, respiratorios y óseos, en particular fracturas espontáneas. Por ello, inicialmente se consideró que podía tratarse de una forma de osteogénesis imperfecta (enfermedad de los huesos de cristal). Sin embargo, la presencia de fibras elásticas fragmentadas permitió incluir esta mutación en la Cutis Laxa. Se trata de una forma recesiva.
EFEMP1 (Fibulina3): Esta mutación provoca múltiples hernias e hipermovilidad articular, además de piel laxa, así como un leve déficit intelectual. Es una nueva forma recesiva de Cutis Laxa.
LTBP1: Con esta mutación se ha observado piel laxa, hernias inguinales, dismorfismo facial, diversos trastornos cardíacos y características esqueléticas marcadas (baja estatura, braquidactilia, craneosinostosis, …). También es una forma recesiva de Cutis Laxa.
PI4K2A: Esta cuarta mutación nueva se caracteriza por los siguientes síntomas clínicos: piel laxa, movimientos involuntarios (trastorno neurológico), dismorfia y retraso intelectual. De nuevo se trata de una forma recesiva.
Recientemente se ha descubierto una quinta mutación nueva y estamos impacientes por que se publique para poder hablar de ella.
Gracias a todos los investigadores por su excepcional trabajo en el conocimiento básico de la Cutis Laxa. Todos estos descubrimientos nos permiten atender mejor a los pacientes y ofrecerles la mejor calidad de vida posible..